
Jacques Teulon

Renal ion channels : from structure to integrated physiology and physiopathology
Ion channel are pore-forming transmembrane proteinsthat control the movement of ions like potassium, chloride and sodium in and out of cells. These movements are critical for a wide-array of renal physiological processes (membrane potential, absorption / secretion, cell volume regulation) and mutations in their encoding genes can lead to disease.
Our group is aimed at integrating ion channels in renal physiology and pathologies. From a better knowledge of the biophysics, regulation and physiological role of renal ion channels, we want to promote their higher integration level in the physiology of the organ and to establish phenotype-genotype relationship for different renal mutations in ion channels-related kidney diseases.
By combining experimental approaches such as single-channel activity in the native renal tissue and cultured cells, functional screening of pathogenic mutations in ion channels, and global in vivo physiological investigation of renal function, we recently produced major contributions in the field of ion channel-related renal diseases:

- The Kir4.1/Kir5.1 protein complex forms the major basolateral K+ conductance of the native distal nephron and a defect in salt re-uptake by distal convoluted tubule (DCT) cells is observed in humans with KCNJ10 mutations, leading to a loss of function of Kir4.1 subunit and ultimately to the hypokalaemic metabolic alcalosis encountered in SeSAME syndrome. We observed that mice with a targeted disruption of the Kcnj16 gene, encoding the Kir5.1 K+ channel regulatory subunit, exhibit a mirror-like image of the SeSAME syndrome, with an increased basolateral K conductance of DCT cells and an intensification of NaCl absorption by the distal convoluted tubule and to hypokalemic metabolic acidosis.

Patch clamp analysis of basolateral K channels in Kir5.1+/+ and Kir5.1-/- mouse DCT cells. From Paulais et al, Proc. Natl. Acad. Sci. U. S. A., 108:10361-10366, 2011.

- A distinct project was devoted to Dent’s disease, an rare X-linked recessive disorder, targeting the proximal tubule, in which we functionally characterized 30 mutations of the ion channel-related 2Cl-/H+ exchanger ClC-5 gene CLCN5 inducing defective processing (Class 1 mutations), delayed processing and decreased stability (Class 2 mutations), and altered ion conduction (Class 3 mutations).

Location of the CLCN5 mutations in a three-dimensional model
of ClC-5 based on the structure of StClC viewed from the side
of the membrane with the extracellular solution at the top.
From Lourdel et al, Pflügers Arch., 463:247-256, 2012

Our current projects are:
Pathophysiological roles of renal K+ channel complexes.
In contrast to their established role along the distal nephron, the function of Kir channels in the proximal nephron, is totally unknown. Our objective is therefore to evaluate their physiological role in the kidney and to assess their possible pathological impact :
1. The cellular location and conductive properties of native Kir channel subunits is currently under investigation using combined molecular (by Western-Blot and
immunohistochemistry) and functional (using the single channel variant of the patch clamp technique) approaches in mouse proximal tubule cells.

Single-channel activity in an isolated mouse renal tubule.
2. In order to determine the impact of dysfunction of Kir channel subunits on proximal ion transport and whole kidney function, we will analyze soon the renal phenotype of Kir knocked-out mice whose breeding is underway in our animal facility.
Pathophysiology, physiology and biophysics of renal ClC-K channels.
Our group has undertaken an exhaustive study of mutations for the CLCNKB gene encoding a Cl- channel, which is implicated in Type III Bartter syndrome, a rare hereditary autosomal recessive renal disease resulting in a salt-wasting tubulopathy associated with hypokalemic alkalosis and secondary hyper aldosteronism. Our past studies showed that all pathogenic CLCNKB mutations alter ClC-Kb channel targeting to the plasma membrane. Still, the properties and regulation of the ClC-Ks are less elucidated than those of other ClC channels. Our current research has two main complementary aspects:
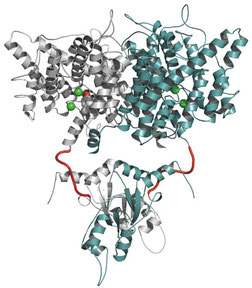
1. For a better understanding of ClC-Ks gating and biophysics, we further investigate the structure-function relationship of ClC-K channels by functional screening of pathogenic and engineered mutations using Xenopus laevis oocytes and cell lines as expression system, electrophysiological techniques, luminescence assays, western-blots, immunofluorescence and design of tagged / untagged ClC-Ks.
Homology model of ClCKb using ClC-ec1 as a template for the membrane domain and ClC- Ka C terminal domain structure (2PFI) for the cytoplasmic domain (Martinez et al, PLoS One, 3 : e2746, 2008).
2. In order to better define ClC-Ks regulation and physiological role, we seek for the impact of intracellular agents and of possible regulatory partners on ClCKs properties using the single-channel and whole-cell variants of the patch-clamp technique in both wild-type and genetically modified mice and on recombinant channels in cell lines.
Team:
Jacques Teulon: Professor UPMC
Marc Paulais : CR1 INSERM
Stéphane Lourdel : MCU UPMC
Olga Andrini : ATER UPMC
Laurent Pinelli : Doctoral student
Representative publications:
2013
L'hoste S, Diakov A, Andrini O, Genete M, Pinelli L, Grand T, Keck M,
Paulais M, Beck L, Korbmacher C, Teulon J, Lourdel S.
Characterization of the mouse ClC-K1/Barttin chloride channel.
Biochim Biophys Acta. 2013 PMID: 23791703
Keck M, Andrini O, Lahuna O, Burgos J, Cid LP, Sepúlveda FV, L'hoste S, Blanchard A, Vargas-Poussou R, Lourdel S, Teulon J. Novel CLCN. KB mutations causing Bartter syndrome affect channel surface expression.Hum. Mutat.In press. PMID:23703872
Le Gars M, Descamps D, Roussel D, Saussereau E, Guillot L, Ruffin M, Tabary O, Hong SS, Boulanger P, Paulais M, Malleret L, Belaaouaj A, Edelman A, Huerre M, Chignard M, Sallenave JM. Neutrophil elastase degrades cystic fibrosis transmembrane conductance regulator via calpains and disables channel function in vitro and in vivo. Am. J. Respir. Crit. Care Med. 187:170-179, 2013. PMID: 23220915
2012
Guinamard R, Paulais M, Lourdel S, Teulon J. A calcium-permeable non-selective cation channel in the thick ascending limb apical membrane of the mouse kidney. Biochim. Biophys. Acta., 1818:1135-1141, 2012. PMID: 22230350
Lourdel S, Grand T, Burgos J, González W, Sepúlveda FV, Teulon J. ClC-5 mutations associated with Dent's disease: a major role of the dimer interface. Pflügers Arch., 463:247-256, 2012. PMID: 22083641
2011
Eladari D, Teulon J. A new regulator of the vacuolar H(+)-ATPase in the kidney. Kidney Int., 80:907-909, 2011. PMID: 21997505
Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel S, Teulon J, Tucker SJ. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc. Natl. Acad. Sci. U. S. A., 108:10361-10366, 2011. PMID: 21633011
Teulon J, Eladari D. A new mouse model for Bartter's syndrome. Am. J. Physiol. Renal Physiol., 301:F295-F296, 2011. PMID: 21632958
Grand T, L'Hoste S, Mordasini D, Defontaine N, Keck M, Pennaforte T, Genete M, Laghmani K, Teulon J, Lourdel S. Heterogeneity in the processing of CLCN5 mutants related to Dent disease. Hum. Mutat., 32:476-483, 2011. PMID: 21305656
2009
Grand T, Mordasini D, L'Hoste S, Pennaforte T, Genete M, Biyeyeme MJ, Vargas-Poussou R, Blanchard A, Teulon J, Lourdel S. Novel CLCN5 mutations in patients with Dent's disease result in altered ion currents or impaired exchanger processing. Kidney Int., 76:999-1005, 2009. PMID: 19657328
2008
Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am. J. Physiol. Renal Physiol., 294:F1398-F1407, 2008. PMID: 18367659